
Inhaltsverzeichnis
Behandeln
Wie wird Hämophilie behandelt?
Da es sich bei Hämophilie um eine Gerinnungsstörung handelt, besteht die aktuelle Standardtherapie darin, den fehlenden Gerinnungsfaktor in Form einer Injektion zu verabreichen.
Übersicht der Hämophilietherapiearten
Der Patient mit Hämophilie hat heutzutage folgende Therapiemöglichkeiten zur Verfügung:
| Therapieansatz | Verabreichung | |
|---|---|---|
| Hämophilie A (Faktor VIII nicht oder zu wenig vorhanden) | Faktorpräparate (Ersatz des Faktor VIII) | Intravenös (in die Armvene gespritzt) |
| Bispezifischer Antikörper (übernimmt die Funktion des Faktor VIII) | Subkutan (direkt unter die Haut gespritzt) | |
| Hämophilie B (Faktor IX nicht oder zu wenig vorhanden) | Faktorpräparate (Ersatz des Faktor IX) | Intravenös (in die Armvene gespritzt) |
Verschaffen Sie sich hier einen Überblick zu den Injektionsarten.
Grundstein der Behandlung: Ersatz für den fehlenden Gerinnungsfaktor
Seitdem bekannt ist, dass bei Hämophilie ein Gerinnungsfaktor teilweise oder gänzlich fehlt und das Blut so bei einer Verletzung nicht gerinnen kann, setzt hier die aktuelle Standardtherapie an: Der fehlende Gerinnungsfaktor wird durch ein Faktorpräparat oder durch einen Antikörper ersetzt. Bei Hämophilie A ist dies der Faktor VIII und bei Hämophilie B ist dies der Faktor IX.
Zu Beginn der sogenannten Faktorersatztherapie in den 1950er Jahren wurde der fehlende Faktor zusammen mit anderen Eiweissen aus menschlichem Blut eines Spenders gewonnen. Dieses Eiweissgemisch, die sogenannte Cohn-Fraktion, erhielt der Hämophilie-Patient bei einer akuten Blutung als intravenöse Injektion. Dadurch konnte die Gerinnungskaskade zu Ende geführt und Blutungen zuverlässig gestoppt werden.
Wenige Jahre später gelang es Wissenschaftlern, den für die jeweilige Blutgerinnungsstörung passenden Faktor einzeln aus menschlichem Blutplasma zu gewinnen: Es war also möglich, den Hämophilie-Patienten nur genau den bei ihnen fehlenden Faktor – Faktor VIII oder Faktor IX – zu verabreichen. Dies wurde als plasmatisches Faktorkonzentrat bezeichnet. Seit nun mehr als 10 Jahren gibt es plasmatische und rekombinante Faktorpräparate auf dem Markt. Die meisten Patienten wenden die Präparate zu Hause an (Heimselbstbehandlung) und müssen nicht mehr regelmässig ins Spital fahren.
Forschung und Entwicklung in der Hämophilie
Das Faktormedikament wurde zunächst aus Spenderblut gewonnen. Deshalb war es zu Beginn noch möglich, dass nicht nur der Faktor, sondern auch Krankheitserreger vom Spender auf den Patienten übertragen werden konnten. Viele Hämophilie-Patienten erkrankten deshalb an Hepatitis B und C. Besonders dramatisch war in den 80er Jahren die Übertragung des HI-Virus durch ein Faktormedikament.
Daraufhin suchte man nach Wegen, um die Faktormedikamente sicherer zu machen. Deshalb wendet man seitdem spezielle Verfahren an um Krankheitserreger – Viren und Bakterien – in den aus Blutplasma hergestellten (plasmatischen) Medikamenten abzutöten oder zu inaktivieren. Durch weitere Forschung war es möglich, den Faktor biotechnologisch und ohne menschliches Blut herzustellen: Seit 2004 sind in der Schweiz rein biotechnologisch hergestellte, so genannte rekombinante Medikamente erhältlich.
Seit 2019 gibt es ein subkutanes Präparat zur prophylaktischen Behandlung von schwerer Hämophilie A. Ein biotechnisch hergestellter, bispezifischer Antikörper übernimmt im Körper die Funktion des fehlenden oder in zu geringen Mengen vorhandenen Faktors VIII und beschleunigt so die Blutgerinnung. Das Präparat wird direkt unter die Haut gespritzt (subkutan).
Prophylaxe oder Bedarfsbehandlung: der Schweregrad entscheidet
Die fehlenden Faktoren werden nicht nur bei einer akuten Behandlung eingesetzt, sondern – je nach Schweregrad der Hämophilie – auch vorbeugend, also prophylaktisch. Bei Patienten mit mittelschwerer und schwerer Hämophilie können so Spontanblutungen reduziert und Gelenkschäden langfristig minimiert werden.
So ist es vielen Patienten heute möglich, mit wenigen Einschränkungen zu leben. Allerdings gibt es dabei folgendes zu beachten: Die Faktorpräparate sollten in regelmässigen Abständen verabreicht werden. Das liegt daran, dass der Faktor in der Leber abgebaut wird. Die Geschwindigkeit, auch Halbwertszeit genannt, mit der er abgebaut wird, ist von Mensch zu Mensch und von Präparat zu Präparat verschieden. Um zu ermitteln, wie häufig ein Präparat verabreicht werden muss, wird die Halbwertszeit des Faktors im Blut ermittelt.
Die Halbwertszeit gibt einen Zeitraum an, in dem sich die Menge des Faktors im Blut auf die Hälfte reduziert. Bei einer Halbwertszeit von zwölf Stunden wäre also zwölf Stunden nach der Verabreichung nur noch die Hälfte der ursprünglichen Faktormenge vorhanden. Nach weiteren zwölf Stunden dann die Hälfte der Hälfte und so weiter. Nach einer bestimmten Zeit ist also nur noch sehr wenig Faktor VIII im Blut vorhanden. Es ist also nötig, neuen Faktor zu verabreichen, damit die Gerinnung weiter funktioniert. Je länger also die Halbwertszeit ist, desto seltener muss der Patient sich das Medikament verabreichen. Eine anschauliche Erklärung der Halbwertszeit bietet das Video der Schweizerischen Hämophilie Gesellschaft.
Inhibitoren erschweren die Behandlung
Eine schwerwiegende Komplikation bei Ersatz eines Gerinnungsfaktors stellt die Entwicklung eines Hemmkörpers dar, eines „Inhibitors“, der die Wirksamkeit des ersetzten Faktors drastisch vermindert.
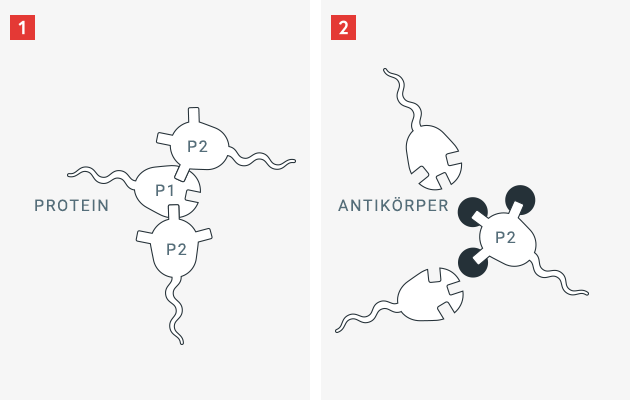
Das Immunsystem des Menschen ist darauf ausgerichtet, ihn vor Gefahren zu schützen, insbesondere vor eindringenden Krankheitserregern wie Bakterien, Viren und Pilze. Um sich gegen diese allgegenwärtigen Eindringlinge zu schützen, hat das Immunsystem die Fähigkeit erworben, fremde Oberflächeneigenschaften von körpereigenen Strukturen und Proteinen zu unterscheiden. Sobald ein Fremdeiweiss für das Immunsystem erkennbar wird, produzieren Abwehrzellen des Körpers nach einer Reihe von Zwischenschritten Gegenabwehrstoffe, sogenannte Antikörper.
Antikörper sind Eiweisse, die sich sehr spezifisch an definierte Oberflächenstrukturen binden können. Durch diese Bindung kann der Körper ein Fremdeiweiss unwirksam machen und es leichter abbauen. Mit körpereigenen Eiweissen (Proteinen) passiert dies nicht, da der menschliche Körper und sein Immunsystem im Rahmen seiner Entwicklung und Reifung den Unterschied zwischen „Fremd“ und „Eigen“ erlernt. Da ein Patient mit einem angeborenen Gerinnungsfaktormangel dieses spezifische Gerinnungsprotein nicht, oder nur in veränderter (und damit weniger wirksamer Form) besitzt, hat das Immunsystem nicht erlernt, dieses normalerweise vorhandene Gerinnungseiweiss als „Eigen“ zu erkennen. Wird dem Patienten das notwendige Gerinnungsprotein (z. B. Faktor VIII) zugeführt, kann es passieren, dass das Immunsystem dieses als „Fremd“ identifiziert – und spezifische Antikörper dagegen bildet.
In der Hämophilie sind diese Antikörper auch unter dem Begriff Inhibitor oder Hemmkörper bekannt. Diese spezifischen Antikörper können das zugeführte Gerinnungseiweiss unwirksam, schwächer wirksam und weniger langlebig werden lassen – sodass der Patient den Schutz vor Blutungen durch die Zufuhr des Medikamentes verliert.
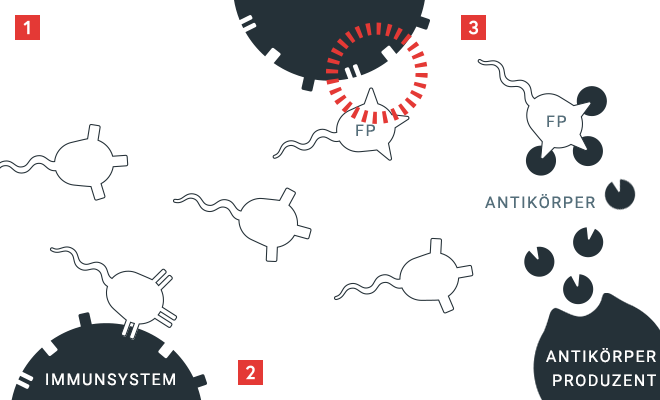
Prinzip der Hemmkörperbildung
Das Immunsystem überprüft Proteine nach ihm vertrauten „Eigen“-Merkmalen. Werden Proteine als „Fremd“ erkannt (FP), werden Antikörper hergestellt, die an diese unbekannten Eigenschaften binden. Bei einer Hemmkörperbildung entstehen Antikörper gegen den gespritzen Faktor VIII – er wird dadurch unwirksam.
Risikofaktoren für eine Inhibitorbildung
Nicht alle Gerinnungsstörungen tragen bei der Substitutionstherapie (Ersatz des fehlenden Faktors) ein gleich hohes Risiko für eine Inhibitorbildung. Das Risiko einer Inhibitorbildung ist etwa während der ersten 50 Einzelgaben am höchsten und sinkt dann bis zur 150. Gabe auf ein stabiles geringes Niveau, welches bestehen bleibt.
Für die Bildung von Inhibitoren gibt es angeborene, also nicht beeinflussbare Risikofaktoren, wie zum Beispiel die ethnische Herkunft oder das Vorhandensein eines Verwandten mit Inhibitoren und erworbene Risikofaktoren – diese sind minimierbar und Gegenstand der Therapieplanung des Arztes mit dem Patienten. Sind Inhibitoren vorhanden und blutet der Patient, so muss dieser weiter behandelt werden, um keinen Schaden durch die Blutung zu nehmen.
Therapeutische Ansätze
Liegt eine Störung der Blutgerinnung mit Inhibitorbildung vor, gibt es heutzutage mehrere therapeutische Möglichkeiten, die es erlauben, die Fibrinproduktion zu aktivieren und somit eine funktionierende Blutgerinnung wiederherzustellen.
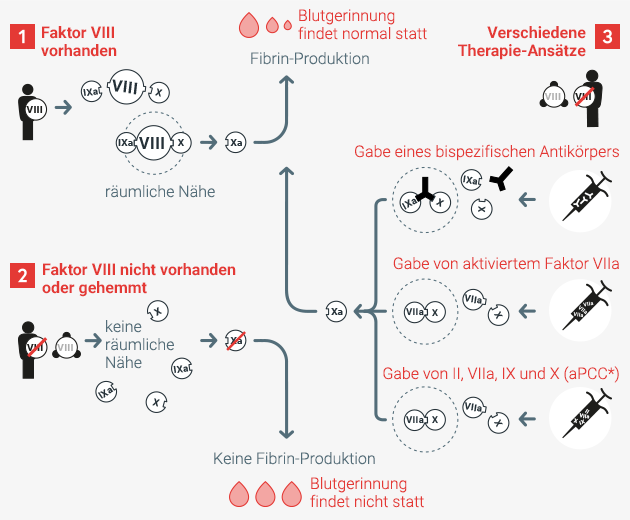
Fall 1
In der gesunden Blutgerinnung sorgt Faktor VIII in engem räumlichen Bezug zu Faktor IX und X für die effektive Herstellung von Xa. Dieser ist für die Fibrinproduktion notwendig, welche das Blut gerinnen lässt.
Fall 2
Ist Faktor VIII nicht vorhanden oder wegen Antikörperbindung nicht aktiv, fehlt der räumliche Bezug von Faktor IXa, VIIIa und X. Xa wird nicht effektiv erzeugt, wodurch die Blutgerinnung gehemmt ist.
Fall 3
Durch Gabe eines bispezifischen Antikörpers bringt der Wirkstoff die Faktoren IXa und X in enge räumliche Nähe, ohne Faktor VIII dafür zu benötigen. Alternativ kann auch die Gabe von aktiviertem Faktor VIIa oder in Kombination mit aPPC (aktiviertes Prothrombinkomplex-Konzentrat) zur Bildung von Faktor VIII führen und somit eine funktionierende Blutgerinnung gewährleisten.
In allen drei Fällen entsteht effektiv Xa und die Blutgerinnung funktioniert.
Es gibt Möglichkeiten einen Inhibitor wieder aus dem Körper zu „verbannen“ – indem das Immunsystem durch eine Therapie erlernt, das zugeführte Gerinnungseiweiss zu tolerieren – man nennt dies „Immuntoleranztherapie“. Diese Therapieform dauert Monate bis Jahre und ist für den Patienten sehr aufwendig. Bei bestimmten Patienten, die eine besondere Inhibitorform aufweisen, wird zusätzlich das Immunsystem medikamentös unterdrückt, um die Bildung von Inhibitoren zu verhindern.
- Schweizerische Hämophilie-Gesellschaft www.shg.ch
- World Federation of Hemophilia www.wfh.org
- Selpers www.selfers.com
- Deutsche Hämophiliegesellschaft www.dhg.de
- Schmerz Nachrichten OÄ Dr. Waltraud Stromer: Hämophilie und Schmerztherapie (pdf)
- Uniklinikum Saarland Dr. med. Susan Halimeh: Die Bedeutung der Physiotherapie in der modernen Hämoph…