
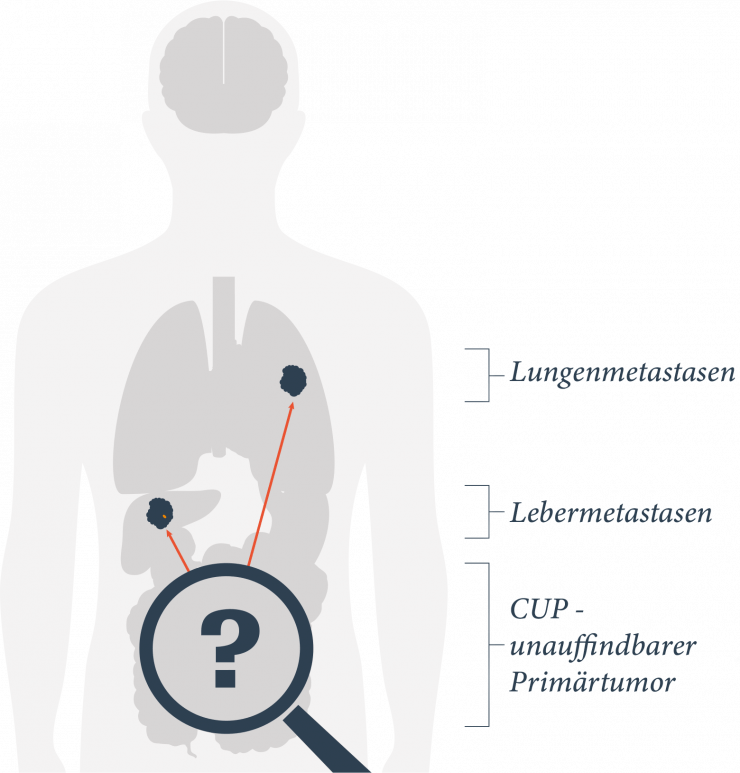
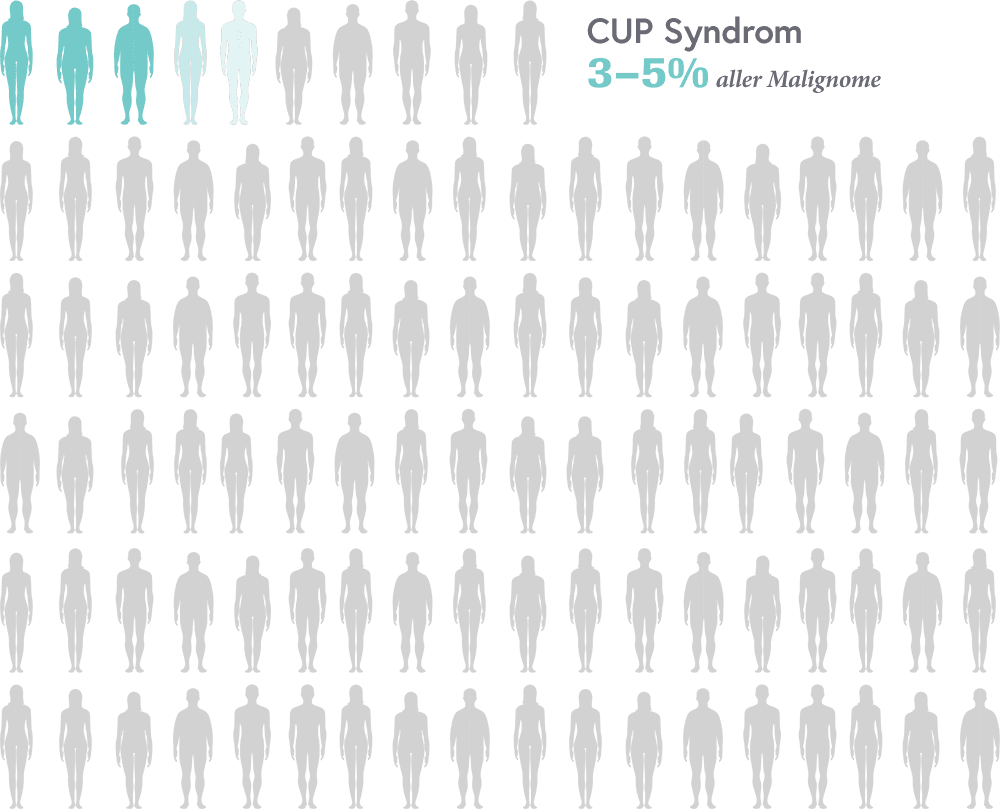
Bildgebende Verfahren
Es gibt verschiedene Verfahren, die in der medizinischen Diagnostik zur Darstellung des Körperinneren verwendet werden:
- Bei der Röntgenuntersuchung wird unsichtbare Strahlung eingesetzt, um den relevanten Körperteil abzubilden
- Bei der Computertomografie (CT) werden Schnittbilder des Körperinneren mithilfe von Röntgenstrahlen erstellt.
- Mit der Magnetresonanztomografie (MRI) werden Schnittbilder des Körperinneren erzeugt, aber ohne Röntgenstrahlung.
- Die Ultraschalluntersuchung oder Sonografie ist ein bildgebendes Verfahren durch den Einsatz von Ultraschall.
- Bei der Endoskopie wird ein Hohlorgan oder eine Körperhöhle (beispielsweise der Bauchraum) mithilfe einer kleinen Kamera visuell von innen untersucht.
Labortests
Labortests an Körperflüssigkeiten wie Blut, Urin usw. können Aufschluss über die Funktion der Organe des Körpers bieten. Der qualitative Nachweis spezifischer Substanzen im Blut (so genannter Biomarker) kann zur Identifizierung des vermutlichen Tumorherds beitragen.
Histologie
Hierbei wird eine Gewebeprobe (das Biopsat) im Labor speziell eingefärbt und unter dem Lichtmikroskop mit einer eingefärbten Probe aus normalen Zellen verglichen. Dadurch lassen sich die Unterschiede zwischen den normalen Zellen und dem Biopsat identifizieren.
Immunhistochemie
Hierbei wird geprüft, ob spezifische Antikörper durch Wechselwirkung mit dem Biopsat zur Identifizierung der genauen Krebsart beitragen können.
Genomtests
Genomtests sind ein Versuch zur Identifizierung der möglicherweise veränderten Gene, die somit zum Wachstum und der Verbreitung des Krebses beigetragen haben. Daraus können sich weitere Behandlungsmöglichkeiten ergeben.
