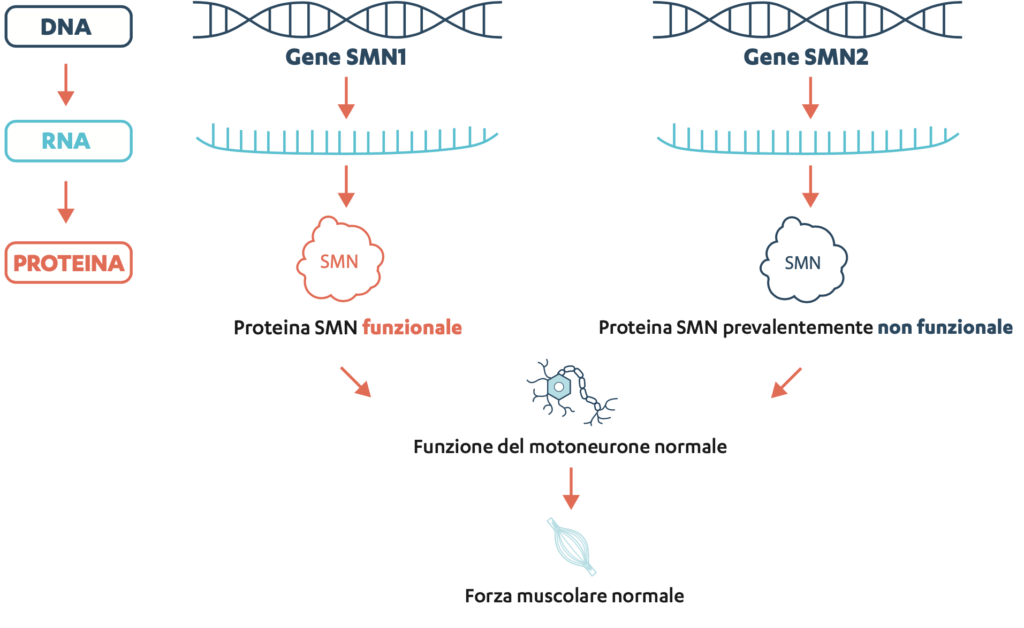
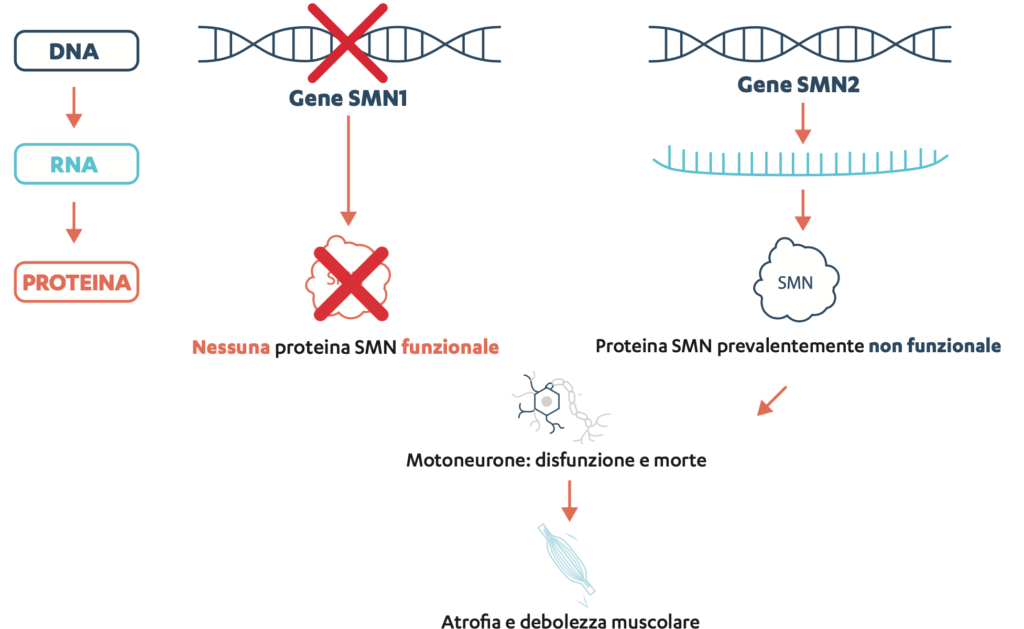









L’atrofia muscolare spinale (SMA) è una malattia rara, progressiva ed ereditaria che causa o può causare debolezza muscolare e altre limitazioni in vari sistemi di organi. Alla base di questa malattia c’è una mancanza o un’alterazione del cosiddetto gene SMN1 nel patrimonio genetico. Questo gene forma il progetto di un’importante proteina, la proteina «Survival of Motor Neuron» (SMN).
La proteina SMN è prodotta ovunque nel corpo ed è necessaria in diverse cellule del corpo. Tuttavia, la proteina SMN è particolarmente importante per la funzione e la sopravvivenza dei motoneuroni, quei neuroni/cellule nervose che sono importanti per il funzionamento dei muscoli. Ecco perché viene denominata anche fattore di protezione dei nervi.
Oltre al gene SMN1, la maggior parte dei soggetti ha anche il gene SMN2, che produce anche la proteina SMN. Se il gene SMN1 manca o è alterato, il gene SMN2 può «subentrare». Tuttavia, il gene SMN2 produce per lo più una proteina tronca e incompleta e solo una piccola quantità di proteina SMN funzionale.
DNA sta per deoxyribonucleic acid, in italiano acido desossiribonucleico, ed è portatore delle informazioni ereditarie in tutti gli esseri viventi. Il DNA è contenuto in ogni cellula del corpo e si trova nel nucleo della cellula.
RNA sta per ribonucleic acid, in italiano acido ribonucleico, e gioca un ruolo chiave nella produzione di proteine, in quanto fornisce le istruzioni per la costruzione delle proteine.
In una persona non affetta da SMA:
In una persona affetta da SMA
In caso di carenza di SMN, i motoneuroni si «ammalano» e alla fine muoiono. I muscoli ricevono sempre meno impulsi nervosi e non possono più funzionare correttamente, si indeboliscono e regrediscono.
La SMA non colpisce solo la muscolatura scheletrica. Oltre ad una mobilità sempre più limitata, le persone affette da SMA possono sviluppare altri sintomi associati o complicazioni come le seguenti.
Problemi a deglutire e mangiare
Debolezza dei muscoli respiratori e difficoltà ad espettorare, che può portare ad una maggiore suscettibilità alla polmonite e ad ulteriori problemi respiratori
Semplici infezioni respiratorie possono portare rapidamente a situazioni di emergenza
Scoliosi (curvatura della colonna vertebrale) e deformità delle ossa
Instabilità dell’anca
Contratture (irrigidimento e restrizione dei movimenti) di articolazioni e muscoli
Problemi gastrointestinali, tra cui reflusso (risalita del contenuto acido dello stomaco nell’esofago), costipazione dovuta alla mancanza di peristalsi
Affaticamento, aumento della stanchezza
Diminuzione della densità ossea, tendenza alle fratture ossee
Il decorso dell’atrofia muscolare spinale varia molto da soggetto a soggetto. Poiché il gene SMN1 non può produrre la proteina SMN nei soggetti affetti da SMA, la quantità di proteina SMN presente dipende in parte da quante copie funzionali del gene SMN2 sono presenti: più sono le copie del gene SMN2 nei soggetti affetti da SMA, maggiore è la proteina SMN funzionale che può essere prodotta.
Anche se con un numero di copie più alto del gene SMN2 la malattia generalmente progredisce più lentamente o è meno grave, al giorno d’oggi non è raccomandata una previsione dell’effettiva gravità della SMA puramente sulla base del numero di copie SMN2, poiché ci possono essere differenze di gravità anche con lo stesso numero di copie.
I passaggi tra i diversi tipi di SMA (tradizionalmente suddivisa dal tipo 0 al tipo 4) sono fluidi e una previsione del corso della malattia (prognosi) non è possibile nei singoli casi solo in base alla classificazione del tipo. Alcuni soggetti affetti mostrano anche sintomi che si collocano tra i tipi descritti. Pertanto, il trattamento dei soggetti affetti da SMA si adatta al rispettivo stato funzionale e ai sintomi. La classificazione avviene quindi in base alle attuali, reali capacità motorie in non-sitter (non in grado di stare seduti in modo autonomo), sitter (in grado di stare seduti in modo autonomo ma non di camminare) e walker (in grado di camminare).
La classificazione del tipo si basa sull’età in cui compaiono per la prima volta i sintomi e il maggiore traguardo raggiunto nello sviluppo fisico.
I sintomi compaiono perlopiù prima dei 6 mesi di età.
Stare seduti non è mai possibile.
I sintomi compaiono perlopiù tra i 6 e i 18 mesi di età. Stare seduti è possibile, stare in piedi non è mai possibile.
I sintomi compaiono generalmente per la prima volta dopo i 18 mesi di età, ma possono comparire tardi nell’infanzia o anche nella prima età adulta. È possibile stare in piedi e camminare.
Inoltre, vi sono due forme meno comuni di SMA:
Malattie oncologiche
Servizio
Disclaimer